Ученые из лаборатории Cold Spring Harbor Laboratory (CSHL) разработали поразительный новый подход к лечению синдрома Ретта – тяжелого расстройства аутистического спектра, которое поражает 1 из 10 000 людей, в основном – девочек.
В статье, выпущенной 27 июля 2015 года на сайте журнала Journal of Clinical Investigation, профессор Николас Тонк и его коллеги продемонстрировали, что лечение при помощи низкомолекулярного препарата значительно увеличивает продолжительность жизни у самцов мышей, у которых моделирован синдром Ретта, и улучшает некоторые поведенческие симптомы заболевания у моделей-самок мышей.
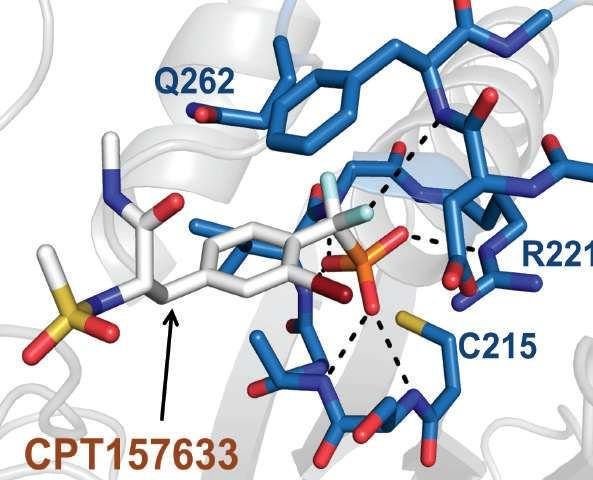
Рентгеновская кристаллография показывает на атомном уровне, как экспериментальный препарат профессора Тонкса при синдроме Ретта, названный CPT157633, связывается со своей мишенью – ферментом PTP1B, который помогает регулировать ключевой метаболический каскад передачи сигналов.
«В настоящее время не существует модифицирующего лечения синдрома, — отмечает доктор Тонкс, — а внимание большинства современных методов терапии сосредоточено на управлении симптомами.
Наш подход представляет собой новую стратегию. В исследовании мы получили четкие доказательства у мышей, что мы можем на самом деле обратить симптомы вспять – это подтверждает концепцию, что заболевание не является необратимым и может поддаваться лечению при помощи низкомолекулярных препаратов».
Синдром Ретта – это наследственное заболевание, связанное с Х-хромосомой, которое нарушает работу головного мозга и нервной системы. Он вызван мутациями в гене под названием MECP2 в Х-хромосоме, который регулирует активность других генов.
Мужчины имеют одну копию Х-хромосомы и одну копию Y-хромосомы. Женщины не имеют Y-хромосомы, а имеют две Х-хромосомы, одна из которых, как правило, инактивируется.
Мальчики, которые рождаются с мутацией MECP2, не имеют другой копии гена, и часто умирают в первый год жизни или два. Девочки с мутацией часто выживают, так как они имеют другую копию гена.
Первый год или два их жизни часто нормален, но в возрасте двух лет начинаются нарушения развития, включая прекращение развития головного мозга, нарушения походки, утрата уже приобретенных социальных навыков. 9 из 10 пациентов с синдромом – это девочки, так как большинство мальчиков не переживают младенческий возраст.
Обнадеживающее исследование, о котором сообщили 27 июля 2015 года, — это продукт 25 лет исследований, проведенных профессором Тонксом.
В 1988 году Николас Тонкс, биохимик, открыл фермент под названием PTP1B, который оказался первым в семействе таких белков, которые в настоящее время насчитывают 105 видов; являясь фосфатазами, эти ферменты удаляют фосфатные группы из других белков.
Эти ферменты являются центральными игроками в каскадах сигналов, посылаемых между клетками и внутри них, для контролирования фундаментальных процессов, таких как рост и метаболизм.
В самом деле, было показано, что PTP1B участвует в подавлении передачи сигналов при помощи гормонов инсулина и лептина, которые, соответственно, занимают центральное место в патологии диабета и ожирения.
Следовательно, препараты, которые ингибируют активность PTP1B, показали потенциал в качестве новых подходов для лечения этих серьезных заболеваний. Выявление и характеристика таких препаратов и было одним из направлений исследования группы профессора Тонкса.
Рисунок показывает, как у здоровых индивидуумов (левая сторона) фактор роста нервных клеток BDNF стимулирует передачу сигналов через рецептор TRKB в клетках головного мозга. Ген MECP2 сохраняет уровни PTP1B низкими, открывая путь.
У пациентов с синдромом Ретта (правая сторона), потеря функции MECP2 приводит к значительному росту уровней, который тормозит путь, при помощи которого в норме BDNF инициирует передачу сигналов через TRKB.
Экспериментальный препарат, разработанный в лаборатории профессора Тонкса, ингибирует PTP1B у мышей, у которых смоделирован синдром, эффективно освобождая торможение передачи сигналов BDNF-TRKB.
После этого наступило обратное развитие симптомов Ретта. Ген MECP2 подавляет экспрессию PTP1B, который усиливает вызванную BDNF передачу сигналов через TRKB протеинтирозинкиназу. Функциональная потеря MECP2 при Ретта приводит к повышению уровней, что ослабляет передачу сигналов BDNF-TRKB.
Осознавая, что метаболическое регулирование, кажется, при синдроме Ретта ненормальное, Навасона Кришнан, научный сотрудник, работающий с профессором Тонксом, предложил использовать ингибиторы PTP1B для того, чтобы изучить, могут ли они повлиять на один из симптомов этой болезни.
Он продемонстрировал ранее, что уровни PTP1B были аномально увеличены у мышиных моделей. Это был обнадеживающий признак того, что ингибиторы PTP1B могут принести пользу при Ретта у мышей. У молодых самцов мышей, лишенных гена Mecp2 (аналог человеческого гена MECP2, который причастен к синдрому), один из ингибиторов PTP1B увеличил среднюю продолжительность жизни от 50 дней до примерно 75 дней. Еще одна молекула работала даже лучше, увеличив выживание до 90 дней.
У более зрелых самок мышей, моделирующих заболевание, ингибиторы PTP1B улучшили симптомы в поведенческих тестах. В одном тесте, прижимание лапок (аналог стереотаксических движений рук у людей) уменьшилось после введения ингибитора PTP1B – частота у леченных мышей снизилась до 25% от примерно 100% до лечения.
А в тесте ходьбы, мыши, моделирующие синдром Ретта, которые помещались на вращающемся колесе, продемонстрировали частичное восстановление функции – они смогли оставаться на колесе без падения значительно дольше, чем мыши, моделирующие болезнь, которым лечение не проводилось.
Профессор Тонкс, доктор Кришнан и их коллеги захотели узнать, были ли за наблюдаемые улучшения ответственны только свойства по регулированию глюкозы ингибиторов PTP1B, которые использовались в экспериментах. Когда мышиным моделям провели лечения двумя основными препаратами при диабете – метформином и розиглитазоном – не наблюдалось никакого улучшения. Это подтвердило, что при введении ингибиторов PTP1B происходит что-то другое. Но что?
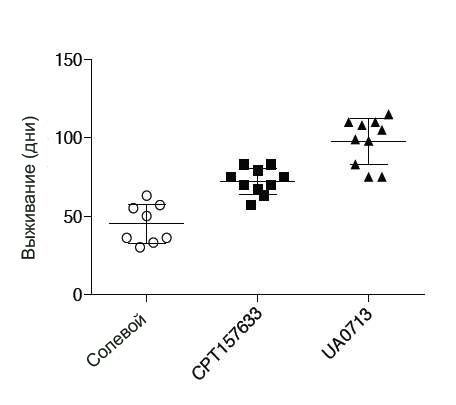
Относительно немного мальчиков, рожденных с мутациями гена MECP2, которые приводят к развитию синдрома Ретта, выживает. У самцов мышей, которые моделируют Ретта, средняя выживаемость увеличилась от примерно 50 дней у животных, которым не проводилось лечение экспериментальными препаратами CPT157633 и UA0713, до примерно 75 и 90 дней, соответственно, после курса лечение каждым из этих соединений.
Дальнейшие эксперименты показали связь междуPTP1B и нейротрофическим фактором роста под названием BDNF, который очень важен для раннего развития головного мозга (и для его дальнейшего функционирования). BDNF взаимодействует с клеточными рецепторами под названием TRKB («track-B»).«В нормальных условиях уровни PTP1B низкие, что способствует передаче сигналов через TRKB, — объясняет профессор Тонкс.
– При синдроме Ретта, уровни PTP1B чрезвычайно высокие, а передача сигналов через TRKB ослаблена». Это на молекулярном уровне подтверждает объяснение, почему ингибиторы PTP1B помогают улучшить симптомы синдрома. «Когда Вы ингибируете PTP1B, как это делают наши препараты, Вы восстанавливаете передачу сигналов BDNF через рецепторы TRKB», — говорит он.
Некоторые нынешние усилия по разработке препаратов стремятся активизировать путь TRKB при синдроме, чтобы восстановить ответы BDNF. Однако, такие подходы не принимают во внимание барьер, который представляют собой высокие уровни PTP1B.
Профессор Тонкс сравнивает это с попыткой езды на машине, нажимая одновременно на педаль газа и тормоза. «Так как PTP1B действует в качестве «тормоза» в модели Ретта, — говорит он, — наш подход подавления PTP1B показал, что произойдет, если Вы просто заберете свою ногу с подали тормоза!»
Лаборатория профессора Тонкса уже начала сотрудничать с другими экспертами в клинических проявлениях синдрома Ретта, чтобы гораздо лучше понять, как ингибирование PTP1B будет модифицировать различные эффекты этой болезни на организм.
Дорогие друзья. Данный материал не является медицинским советом, за диагнозом и способом лечения, обратитесь к специалисту для консультации.
