Наследственный фактор лежит в основе развития многих заболеваний, как психических, душевных, так и соматических, телесных.
Чем раньше выявлена передавшаяся по наследству патология, тем раньше можно провести лечение. Соответственно, больше шансов на частичное или даже полное восстановление.
Содержание
Общие принципы
Все соматические, и даже психические функции представляют собой совокупность обменных или метаболических процессов. Эти процессы, в свою очередь, регулируются эндокринной системой и нервной, центральной и периферической, системой.
Каждый физиологический процесс посредством ферментов или энзимов контролируется одним или несколькими генами. Аналогичные парные гены наследуются от обоих родителей, и называются аллелями. Они влияют на структуру органов и тканей. В общем, генотип влияет на фенотип. От совокупности аллелей зависят фенотипические, индивидуальные характеристики человека.
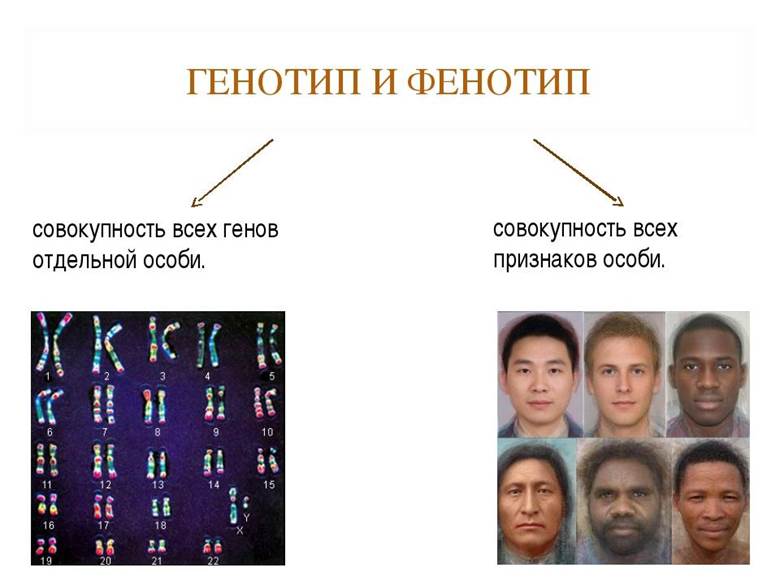
Генетические мутации часто приводят к изменениям фенотипа, к нарушениям нейроэндокринной регуляции, к метаболическим сдвигам. Все это проявляется патологиями. Поскольку мы наследуем гены от родителей, то и болезни, развившиеся из-за возникших аномалий, тоже передаются нам.
Виды
Наследственную болезнь часто путают с врожденной. Это не одно и то же. Врожденные не всегда наследуемые. Типичный пример: ребенок зачат генетически, психически, и соматически здоровыми родителями.
Его хромосомный аппарат тоже нормальный. Но во время беременности плод подвергся действию лекарств, инфекции, токсинов, других неблагоприятных факторов, и младенец родился с аномалиями.
Но если такой малыш в будущем доживет до половозрелого возраста, и его репродуктивная система будет сохранена, то дети, скорее всего, будут нормальными. Такие ненаследственные изменения, которые имеют сходство с наследственными, называют фенокопиями.
Примеры фенокопий: заячья губа, волчья пасть, и другие врожденные уродства. В некоторых клинических случаях они имеют наследственный характер, хотя могут развиваться при воздействии неблагоприятных факторов во внутриутробном периоде.
Что касается наследственной, она не всегда проявляется при рождении. Многие патологии дебютируют, впервые заявляют о себе, в раннем детском, подростковом периоде, и даже во взрослом возрасте.
Так, эпилепсия, шизофрения, чаще всего дебютируют в молодые годы. От родителей к детям часто передаются не сами заболевания, а предрасположенность к ним.
Наследуется предрасположенность ко многим соматическим: сердечно-сосудистым, онкологическим, эндокринным. Родители передают детям даже склонность к алкоголизму.
Ведь в основе алкогольной зависимости лежат метаболические нарушения с замедленным расщеплением спирта этилового (этанола). Причина – дефицит расщепляющего этанол фермента алкогольдегидрогеназы из-за мутаций.
Виды генетических отклонений
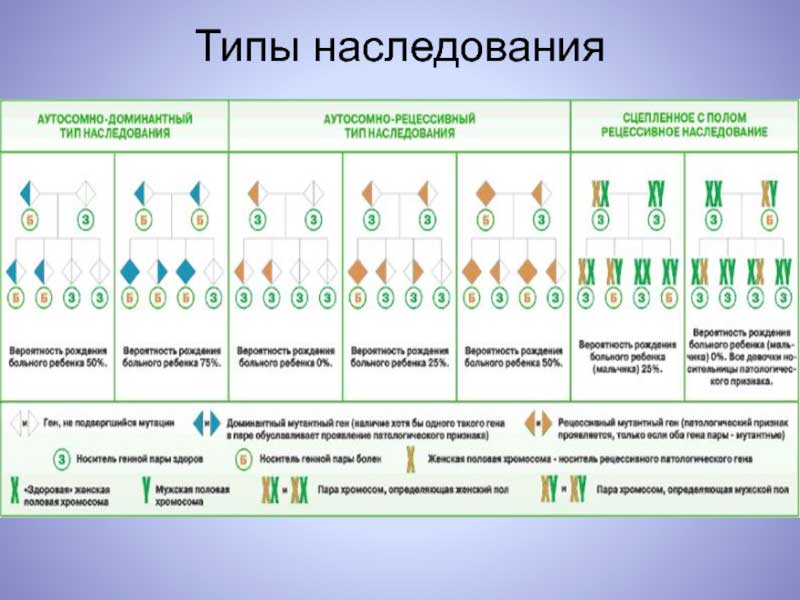
В зависимости от того, на каком уровне возникли мутации, выделяют геномные, хромосомные, и генные врожденные и наследственные аномалии.
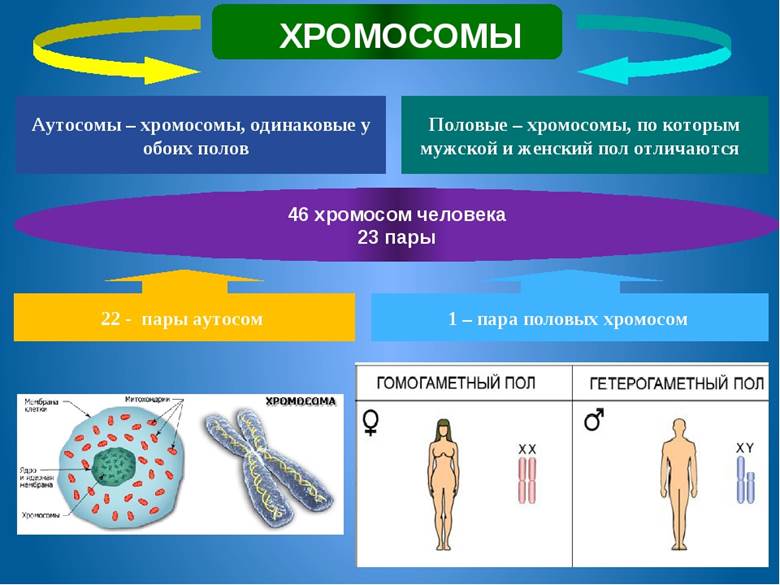
Хромосомная обусловлена изменением структуры отдельных хромосом или их общего числа.
Хромосомный набор или кариотип человека включает 46 хромосом, 23 пары, по каждой от отца и матери в паре. Это 22 пары аутосом, неполовых, и 1 пара половых.
Геномные аномалии характеризуются изменениями на уровне кариотипа. Яркий пример таких аномалий: синдром Дауна, 47 хромосом из-за «лишней» в 21 паре. Геномные мутации, как правило, проявляются уже при рождении, сильно выражены, и порой несовместимы с жизнью.
Хромосомные мутации затрагивают одну хромосому. Это отрыв участка нити ДНК, перенос или дислокация участка, или поворот на 1800. Измененные участки включают большое количество аллелей. Поэтому такие аномалии тоже проявляются тяжелыми расстройствами.
Генные мутации затрагивают точечные участки, локусы, хромосом. Каждому локусу соответствует определенный ген.
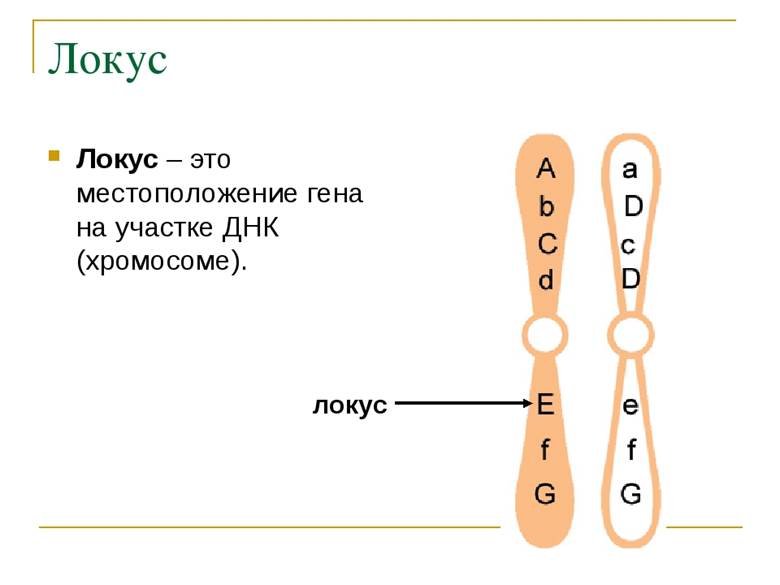
Мутации могут затрагивать один ген, их называют моногенными. Примеры моногенной: синдром Марфана, фенилкетонурия, гемофилия А.
Моногенные аномалии называют еще менделирующими. Они подчиняются законам, выведенным основоположником генетики Менделем в опытах над горохом.
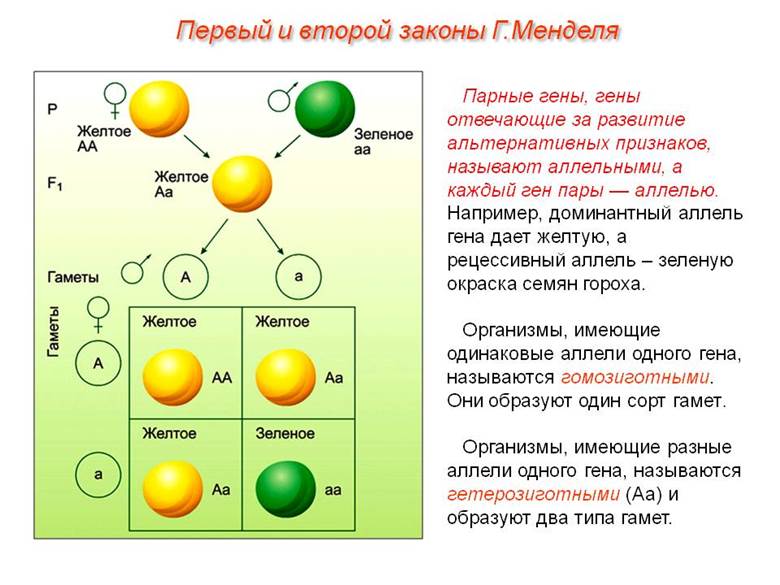
Любопытный факт: многие мутации имеют рецессивный характер. Это значит, что носители измененных генов часто имеют нормальный фенотип.
Попросту говоря, это внешне здоровые люди, если вторая, доминантная, аллель в паре у них нормальная. Чтобы патология проявилась у потомства, нужно, чтобы оба родителя являлись носителями патологически измененного гена.
Вот почему браки между близкими родственниками крайне нежелательны. Дети от таких браков часто страдают наследственными недугами, а иногда вовсе нежизнеспособны. Напротив, многие потомки от меж расовых браков не только внешне красивы, но и здоровы, умны, сильны и выносливы.
При полигенных заболеваниях поражается большое количество генов. Причем измененные аллели находятся не в одной, а в разных хромосомах. Некоторые полигенные мутации заявляют о себе умственной отсталостью, метаболическими изменениями, другими пороками развития уже при рождении или в первые годы жизни.
Но чаще всего они дебютируют в зрелом, или даже в пожилом возрасте. Так по наследству передаются некоторые виды сахарного диабета, гипертония, онкология, эпилепсия, шизофрения, болезнь Альцгеймера, и другие психические расстройства.
Точнее, передаются не сами заболевания, а предрасположенность к ним. Такие реализуются при воздействии на организм психических нагрузок, загрязненной воды и атмосферы, при плохом питании, сопутствующих заболеваниях, и других неблагоприятных факторах.
Один и тот же фактор с большей вероятностью приведет к патологии при наличии мутаций, чем при их отсутствии. Поэтому полигенные нарушения еще называют мультифакторными, т.к. их развитие зависит не только от состояния хромосомного аппарата, но и от множества других причин.
Виды диагностики
Для выявления наследственных прибегают к различным методам.
- Биохимические анализы
Это обширная группа исследований для выявления различных видов изменений метаболизма. В ходе биохимических исследований определяют уровень глюкозы, общего белка, фракций билируби6на и холестерина, другие базовые показатели. При необходимости проводят более углубленные исследования на гормоны, электролиты плазмы крови, специфические ферменты.
- Иммунологические анализы
Исследуют кровь на наличие специфических антител-иммуноглобулинов. Эти антитела продуцируются В-лимфоцитами, и могут блокировать ферменты и гормоны. Нарушение метаболизма сопровождается сбоем в работе органов и систем, накоплением токсических продуктов.
- Цитогенетические анализы
Данный вид используют для выявления мутаций на уровне отдельных хромосом или всего кариотипа. Суть исследования заключается в окрашивании клеточного материала в составе образцов тканей.
Клетки окрашивают по специальной методике, и рассматривают под микроскопом. При микроскопии выявляют патологически измененные участки хромосом, определяют их общее количество.
Для диагностики патологии, сцепленной с полом, прибегают к исследованию полового хроматина, свернутой уплотненной неактивной Х-хромосомы в промежутках между клеточными делениями. Половой хроматин хорошо виден при окрашивании и микроскопии.
- Молекулярно-генетические исследования
Цель этих исследований – определение последовательности аминокислотных или нуклеотидных звеньев в цепях белков, нуклеиновых кисло ДНК и РНК. Самый популярный метод – ПЦР, полимеразная цепная реакция.
Исследование проводится in vitro, в пробирке, и требует дорогостоящей аппаратуры. Суть его заключается в многократном копировании и дальнейшем изучении исследуемого фрагмента ДНК.
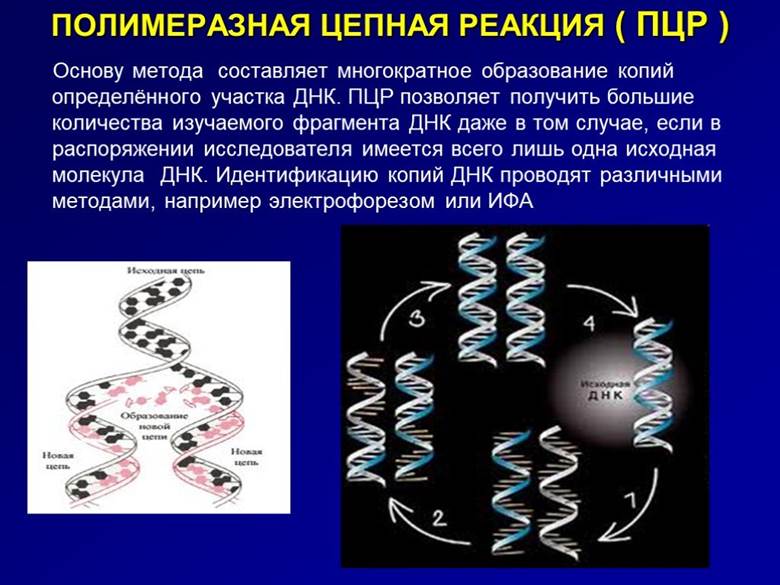
Наряду с ПЦР прибегают к сходным методам: FISH или флуоресцентной гибридизации, микрочипированию.

ПЦР методы отличаются высокой чувствительностью. Достоверность результатов здесь составляет 99%. Помимо исследования генома и генно-хромосомных аномалий их используют для выявления инфекционных и соматических патологий, в криминалистике.
Что можно выявить
Уже в первые дни жизни осуществляют скрининг новорожденных на наличие метаболических расстройств из-за наследственных отклонений. Скрининг – это просеивание, в данном случае – выделение больных детей в отдельные группы для дальнейшей углубленной диагностики и лечения.
Неонатальный скрининг, обследование новорожденных, проводят в условиях родильного отделения на 4-10 день жизни. Материалом для биохимических анализов служит кровь младенца. Ее получают путем прокола пятки ребенка. Поэтому анализы еще называют пяточным тестом. Результат обычно готов через 3-14 дней после взятия материала.
В рамках обязательного скрининга проводят осмотьр 5 врожденных недугов.
- Гипотиреоз
Это недостаточность щитовидной железы, в данном случае врожденного характера. Дефицит щитовидных гормонов у детей проявляется кретинизмом с нарушением всех основных видов метаболизма, отставанием в физическом и психическом развитии. Для гипотиреоза исследуют уровень щитовидного гормона тироксина (Т4) и тиреотропного гормона (ТТГ) гипофиза.
- Адреногенитальный синдром
Врожденный дефицит надпочечникового гормона кортизола. Причина: наследуемый по аутосомно-доминантному типу дефект гена, контролирующего синтез стероид-21-гидроксилазы. Этот фермент регулирует обмен стероидных половых и надпочечниковых гормонов.
Малое кличество этих гормонов сопровождается нарушением водно-солевого обмена, других видов метаболизма, аномальным формирования гениталиев. В рамках скрининговой диагностики проверяют уровень 17-гидроксипрогестерона. При адреногенитальном синдроме уровень этого стероидного гормона повышен.
- Фенилкетонурия
В норме поступающая с пищей аминокислота фенилаланин под действием печеночного фермента фенилаланин-4-гидроксилазы превращается в аминокислоту тирозин. Из тирозина образуется пигмент меланин, гомоны щитовидной железы, а также адреналин, норадреналин.
Недостаток фенилаланин-4-гидроксилазы наследуется по аутосомно-рецессивному типу. При этом в организме формируется избыток фенилаланина и его токсических производных. Болезнь протекает с поражением головного мозга, сердечно-сосудистой системы. Неонатальная направлена на выявление высокого уровня фенилаланина.
- Муковисцидоз
Эта генетическая патология наследуется по аутосомно-рецессивному типу. Характеризуется мутациями гена, расположенного в 7 хромосоме. Этот ген контролирует синтез белка трансмембранного регулятора муковисцидоза, ответственного за перенос ионов хлора через клеточные мембраны. Муковисцидоз протекает с разной степени тяжести поражением органов дыхания и желудочно-кишечного трата. При муковисцидозе уровень иммунореактивного трипсина в крови новорожденного повышен.
- Галактоземия
Галактоза – углевод, поступающий к нам с пищевыми продуктами. Малыши получают его с материнским молоком. В норме галактоза при участии специфических ферментов трансформируется в глюкозу.
Дефицит этого фермента вследствие дефектов приводит к накоплению галактозы и ее токсических производных. Поражается нервная система, печень, почки, желудочно-кишечный тракт, глаза. В крови при неонатальном скрининге выявляют высокий уровень галактозы.
Лабораторная диагностика позволяет выявить несколько десятков наследуемых изменений с метаболическими нарушениями, поражением головного мозга и внутренних органов. Но обязательный неонатальный скрининг включает лишь 5 вышеуказанных изменений как самых актуальных. Их частота составляет 1 случай на 5-30 тыс. новорожденных.
Остальные болезни встречаются еще реже. Но при желании родителей или по медицинским показаниям они тоже могут быть включены в неонатальную.
Расширенный скрининг новорожденных включает в себя большое количество патологий:
| Группа | Заболевания |
| Органические кислоты. | Пропионовая ацидемия. Изовалериановая ацидемия. Метилмалоновая ацидемия. Недостаток ?-кетотиолазы. Дефицит синтетазы холокарбоксилаз. Глутаровая ацидемия 1 типа. 3-гидрокси-3 метилглутаровая ацидурия. Дефицит 3-метилкротонил-коэнзим А карбоксилазы. |
| Жирные кислоты. | Бедность длинноцепочечной 3-гидроксиацил-КоА дегидрогеназы. Дефицит ацил-КоА-дегидрогеназы. Нехватка переносчика карнитина. |
| Аминокислоты. | Фенилкетонурия. Гомоцистеинурия. Лейциноз (болезнь кленового сиропа. Аргининосукциновая ацидурия. Цитруллинемия 1 типа. Тирозинемия 1 типа. |
| Гемоглобинопатии. | Серповидноклеточная анемия. S ? талассемия. Заболевание гемоглобина S-C. |
| Прочие. | Муковисцидоз. Мукополисахаридоз 1 типа. Врожденный гипотиреоз. Адреногенитальный синдром. Болезнь Помпе. Недостаток биотинидазы. Галактоземия. Спинальная мышечная атрофия. Х-сцепленная адренолейкодистрофия. Тяжелый комбинированный иммунодефицит. |
При положительных результатах скрининг-тестов расширяют диагностику. Проводят биохимические, иммунологические исследования. После этого переходят к ПЦР анализам.
Что еще можно определить
С помощью генетических тестов можно выявить предрасположенность к самым распространенным изменениям, сердечно-сосудистым. А поскольку сердечно-сосудистая патология является ведущей причиной смерти, то здесь с большой долей вероятности можно прогнозировать продолжительность жизни обследуемого.
Всем известен холестерин как главный «виновник» в развитии болезней сердца и сосудов. Но не каждому известны позитивные свойства этого липидного, жироподобного соединения. Холестерин участвует в обменных процессах, является сырьем для многих гормонов. Из него состоят клеточные мембраны.
Доставка холестерина к тканям осуществляется не в свободном виде, а в виде липопротеинов (ЛПП), белково-жировых соединений. Липопротеины имеют разную плотность. Чем она ниже, тем эти соединения опаснее. Из липопротеинов низкой и очень низкой плотности (ЛПНП, ЛПОНП) состоят атеросклеротические бляшки, закупоривающие сосуды.
Свойства ЛПП зависят от белкового компонента, аполипопротеина (АРО). Выделяют несколько видов этих белков. Аполипопротеин Е (АРО-Е) входит в состав ЛПНП. Этот белок синтезируется клетками печени и центральной нервной системы (ЦНС). АПО-Е захватывает липидные соединения (триглицериды, холестерин) в плазме крови, и транспортирует их к клеткам тканей.
Таким образом, АРО-Е осуществляет обмен липидов, регулирует их концентрацию в крови. Белок этот неоднороден, и представлен тремя разновидностями или изоформами:
- АРО-Е2
- АРО-Е3
- АРО-Е4.
Изоформы отличаются друг от друга последовательностью и набором аминокислот на определенных участках. Их роль в липидном обмене тоже неодинакова. Лучше всего с поставленными задачами справляется АРО-Е2.
В то же время АРО-Е4 ответственный за отложение жиров в виде атеросклеротических бляшек. Наличие той или иной изоформы АРО-Е предопределено генетически. Синтез этого белка закодирован геном, расположенным в 19 паре хромосом.
От того, какой аллелью представлен этот ген, зависит изоформа АРО-Е. Поскольку каждую аллель в паре хромосом мы получаем от одного из родителей, то здесь возможны несколько вариантов:
- Е2/Е2
- Е2/Е3
- Е3/Е3
- Е3/Е4
- Е4/Е4.
При наличии аллели Е4, унаследованной хотя бы от одного из родителей, повышается риск атеросклероза, ожирения, и связанных с ними болезней: гипертония, ишемической патологии сердца, инфаркта миокарда, мозгового инсульта. Это самые частые причины смертности и инвалидности.
При варианте Е4/Е4, когда аллель наследуется от обоих родителей, риск этих заболеваний максимален. Казалось бы, самым благоприятным является вариант Е2/Е2. Но нет. Если Е2 наследуется от обоих родителей, есть вероятность гиперлипопротеинемии 3 типа (ГЛП-3).
Правда, это тяжело протекающее заболевание с нарушением липидного обмена встречается редко. Таким образом, самым благоприятным является вариант Е3/Е3. И такой вариант самый частый – он встречается у 60% людей.
В некоторых публикациях можно встретить, что ген АРО-Е якобы отвечает за удаление ртути из ЦНС. Ртуть попадает в организм в составе консервантов прививок. И если у ребенка АРО-Е 4, значит, из мозга ртуть вообще не удаляется, и после прививок у него разовьется слабоумие, аутизм, или другие психические расстройства.
Что движет сторонниками этой теории, искреннее заблуждение, или стремление намеренно исказить информацию, сказать трудно. Ясно только, что это чушь полнейшая, как и роль ртути из прививок в развитии детской мозговой патологии.
Кстати о слабоумии. Действительно, при АРО-Е4 вероятность прогрессирования старческого слабоумия, болезни Альцгеймера, выше, чем при других изоформах. Были обследованы дети до 8 лет с аллелью Е4. Установлено, что обучаемость у них ниже, чем у сверстников. Однако по мере взросления когнитивные функции у таких детей восстанавливаются.
Мозговые дисфункции в старческом и в детском возрасте при наличии Е4 связаны с замедленным удалением продуктов липидного обмена из мозговых тканей. В любом случае ясно, что ртуть и прививки здесь не при чем.
Выводы
Несмотря на высокую информативность, анализы не всегда позволяют определить конкретную причину изменений. Ведь один и тот же фермент часто кодируется большим числом генов. Мутации любого такого гена проявляются заболеванием.
Такие же трудности при определении предрасположенности к соматическим и психическим расстройствам. Эти расстройства тоже часто имеют полигенный характер.
Вероятность их возникновения зависит не только от характера дефектов, но и от того, как, насколько интенсивно, действуют внешние неблагоприятные факторы. Поэтому говорить о том, разовьется ли данная патология у конкретного человека на основе результатов генетических тестов можно лишь приблизительно.
Дорогие друзья. Данный материал не является медицинским советом, за диагнозом и способом лечения, обратитесь к специалисту для консультации.
